%matplotlib inline
Data analysis with pynapple & nemos#
This notebook has had all its explanatory text removed and has not been run. It is intended to be downloaded and run locally (or on the provided binder) while listening to the presenter’s explanation. In order to see the fully rendered of this notebook, go here
Learning objectives#
Loading a NWB file
Compute tuning curves
Decode neural activity
Compute correlograms
Include history-related predictors to NeMoS GLM.
Reduce over-fitting with
Basis.Learn functional connectivity.
The pynapple documentation can be found here.
The nemos documentation can be found here.
Let’s start by importing all the packages.
If an import fails, you can do !pip install pynapple nemos matplotlib in a cell to fix it.
import pynapple as nap
import matplotlib.pyplot as plt
import workshop_utils
import numpy as np
import nemos as nmo
# some helper plotting functions
from nemos import _documentation_utils as doc_plots
import workshop_utils
# configure pynapple to ignore conversion warning
nap.nap_config.suppress_conversion_warnings = True
# configure plots some
plt.style.use(nmo.styles.plot_style)
Loading a NWB file#
Pynapple commit to support NWB for data loading. If you have installed the repository, you can run the following cell:
path = workshop_utils.fetch_data("Mouse32-140822.nwb")
print(path)
Pynapple provides the convenience function nap.load_file for loading a NWB file.
Question: Can you open the NWB file giving the variable path to the function load_file and call the output data?
data = nap.load_file(path)
print(data)
The content of the NWB file is not loaded yet. The object data behaves like a dictionnary.
Question: Can you load the spike times from the NWB and call the variables spikes?
spikes = data["units"] # Get spike timings
Question: And print it?
print(spikes)
There are a lot of neurons. The neurons that interest us are the neurons labeled adn.
Question: Using the slicing method of your choice, can you select only the neurons in adn that are above 2 Hz firing rate?
spikes = spikes[(spikes.location=='adn') & (spikes.rate>2.0)]
print(len(spikes))
The NWB file contains other informations about the recording. ry contains the value of the head-direction of the animal over time.
Question: Can you extract the angle of the animal in a variable called angle and print it?
angle = data["ry"]
print(angle)
But are the data actually loaded … or not?
Question: Can you print the underlying data array of angle?
Data are lazy-loaded. This can be useful when reading larger than memory array from disk with memory map.
print(angle.d)
The animal was recorded during wakefulness and sleep.
Question: Can you extract the behavioral intervals in a variable called epochs?
epochs = data["epochs"]
print(epochs)
NWB file can save intervals with multiple labels. The object IntervalSet includes the labels as a metadata object.
Question: Using the column tags, can you create one IntervalSet object for intervals labeled wake and one IntervalSet object for intervals labeled sleep?
wake_ep = epochs[epochs.tags=="wake"]
sleep_ep = epochs[epochs.tags=="sleep"]
Compute tuning curves#
Now that we have spikes and a behavioral feature (i.e. head-direction), we would like to compute the firing rate of neurons as a function of the variable angle during wake_ep.
To do this in pynapple, all you need is a single line of code!
Question: can you compute the firing rate of ADn units as a function of heading direction, i.e. a head-direction tuning curve and call the variable tuning_curves?
tuning_curves = nap.compute_tuning_curves(
data=spikes,
features=angle,
bins=61,
epochs = angle.time_support,
range=(0, 2 * np.pi),
feature_names = ["Angle"]
)
tuning_curves
Question: Can you plot some tuning curves?
plt.figure()
plt.subplot(221)
tuning_curves[0].plot()
# plt.plot(tuning_curves[0])
plt.subplot(222,projection='polar')
plt.plot(tuning_curves.Angle, tuning_curves[0].values)
plt.subplot(223)
tuning_curves[1].plot()
plt.subplot(224,projection='polar')
plt.plot(tuning_curves.Angle, tuning_curves[1].values)
plt.tight_layout()
Most of those neurons are head-directions neurons.
The next cell allows us to get a quick estimate of the neurons’s preferred direction. Since this is a lot of xarray wrangling, it is given.
pref_ang = tuning_curves.idxmax(dim="Angle")
# pref_ang = tuning_curves.idxmax()
Question: Can you add it to the metainformation of spikes?
spikes['pref_ang'] = pref_ang
spikes
This index maps a neuron to a preferred direction between 0 and 360 degrees.
Question: Can you plot the spiking activity of the neurons based on their preferred direction as well as the head-direction of the animal?
For the sake of visibility, you should restrict the data to the following epoch : ex_ep = nap.IntervalSet(start=8910, end=8960).
ex_ep = nap.IntervalSet(start=8910, end=8960)
plt.figure()
plt.subplot(211)
plt.plot(angle.restrict(ex_ep))
plt.ylim(0, 2*np.pi)
plt.subplot(212)
plt.plot(spikes.restrict(ex_ep).to_tsd("pref_ang"), '|')
Decode neural activity#
Population activity clearly codes for head-direction. Can we use the spiking activity of the neurons to infer the current heading of the animal? There are currently 2 ways to do this in pynapple :
bayesian decoding
template matching
Question: Using the method of your choice, can you compute the decoded angle from the spiking activity during wakefulness?
decoded, proba_feature = nap.decode_bayes(
tuning_curves=tuning_curves,
data=spikes,
epochs=wake_ep,
bin_size=0.3, # second
)
# decoded, proba_feature = nap.decode_template(
# tuning_curves=tuning_curves,
# data=spikes,
# epochs=wake_ep,
# bin_size=0.3, # second
# )
Question: … and display the decoded angle next to the true angle?
plt.figure()
plt.subplot(211)
plt.plot(angle.restrict(ex_ep))
plt.plot(decoded.restrict(ex_ep), label="decoded")
plt.ylim(0, 2*np.pi)
plt.subplot(212)
plt.plot(spikes.restrict(ex_ep).to_tsd("pref_ang"), '|')
Since the tuning curves were computed during wakefulness, it is a circular action to decode spiking activity during wakefulness. We can try something more interesting by trying to decode the angle during sleep.
Question: Can you instantiate an IntervalSet object called rem_ep that contains the epochs of REM sleep? You can check the contents of the NWB file by doing first print(data)
rem_ep = data['rem'][1]
Question: Can you compute the decoded angle from the spiking activity during REM sleep?
decoded, proba_feature = nap.decode_bayes(
tuning_curves=tuning_curves,
data=spikes,
epochs=rem_ep,
bin_size=0.3, # second
)
# decoded, proba_feature = nap.decode_template(
# tuning_curves=tuning_curves,
# data=spikes,
# epochs=rem_ep,
# bin_size=0.3, # second
# )
Question: … and display the decoded angle next to the spiking activity?
plt.figure()
plt.subplot(211)
plt.plot(decoded.restrict(rem_ep), label="decoded")
plt.ylim(0, 2*np.pi)
plt.subplot(212)
plt.plot(spikes.restrict(rem_ep).to_tsd("pref_ang"), '|')
Compute correlograms#
We see that some neurons have a correlated activity. Can we measure it?
Question: Can you compute cross-correlograms during wake for all pairs of neurons and call it cc_wake?
cc_wake = nap.compute_crosscorrelogram(spikes, binsize=0.2, windowsize=20.0, ep=wake_ep)
Question: can you plot the cross-correlogram during wake of 2 neurons firing for the same direction?
index = spikes.keys()
plt.figure()
plt.subplot(121)
tuning_curves.sel(unit=[7,20]).plot(x='Angle', hue='unit')
plt.title("Tuning curves")
plt.subplot(122)
plt.plot(cc_wake[(7, 20)])
plt.xlabel("Time lag (s)")
plt.title("Cross-corr.")
Question: can you plot the cross-correlogram during wake of 2 neurons firing for opposite directions?
index = spikes.keys()
plt.figure()
plt.subplot(121)
tuning_curves.sel(unit=[7,26]).plot(x='Angle', hue='unit')
plt.title("Tuning curves")
plt.subplot(122)
plt.plot(cc_wake[(7, 26)])
plt.xlabel("Time lag (s)")
plt.title("Cross-corr.")
Pairwise correlation were computed during wakefulness. The activity of the neurons was also recorded during sleep.
Question: can you compute the cross-correlograms during sleep?
cc_sleep = nap.compute_crosscorrelogram(spikes, 0.02, 1.0, ep=sleep_ep)
Question: can you display the cross-correlogram for wakefulness and sleep of the same pairs of neurons?
plt.figure()
plt.subplot(131)
tuning_curves.sel(unit=[7,20]).plot(x='Angle', hue='unit')
plt.title("Tuning curves")
plt.subplot(132)
plt.plot(cc_wake[(7, 20)])
plt.xlabel("Time lag (s)")
plt.title("Wake")
plt.subplot(133)
plt.plot(cc_sleep[(7, 20)])
plt.xlabel("Time lag (s)")
plt.title("Sleep")
plt.tight_layout()
plt.figure()
plt.subplot(131)
tuning_curves.sel(unit=[7,26]).plot(x='Angle', hue='unit')
plt.title("Tuning curves")
plt.subplot(132)
plt.plot(cc_wake[(7, 26)])
plt.xlabel("Time lag (s)")
plt.title("Wake")
plt.subplot(133)
plt.plot(cc_sleep[(7, 26)])
plt.xlabel("Time lag (s)")
plt.title("Sleep")
plt.tight_layout()
What does it mean for the relationship between cells here?
Fitting a GLM model with Nemos#
wake_ep = nap.IntervalSet(
start=wake_ep.start[0], end=wake_ep.start[0] + 3 * 60
)
To use the GLM, we need first to bin the spike trains. Here we use pynapple and the function count.
Question: can you bin the spike trains in 10 ms bins during the wake_ep and call the variable count?
bin_size = 0.01
count = spikes.count(bin_size, ep=wake_ep)
print(count.shape)
Question: can you reorder the columns of count based on the preferred direction of each neuron?
Above we defined pref_ang as the preferred direction of each neuron.
count = count[:, np.argsort(pref_ang.values)]
It’s time to use NeMoS. Our goal is to estimate the pairwise interaction between neurons. This can be quantified with a GLM if we use the recent population spike history to predict the current time step.
To simplify our life, let’s see first how we can model spike history effects in a single neuron. The simplest approach is to use counts in fixed length window \(i\), \(y_{t-i}, \dots, y_{t-1}\) to predict the next count \(y_{t}\).
Before starting the analysis, let’s
- select a neuron from the count object (call the variable neuron_count)
- Select the first 1.2 seconds for visualization. (call the epoch epoch_one_spk).
# select a neuron's spike count time series
neuron_count = count[:, 0]
# restrict to a smaller time interval
epoch_one_spk = nap.IntervalSet(
start=count.time_support.start[0], end=count.time_support.start[0] + 1.2
)
Features Construction#
Let’s fix the spike history window size that we will use as predictor.
Question: Can you:
Fix a history window of 800ms (0.8 seconds).
Plot the result using
doc_plots.plot_history_window
# set the size of the spike history window in seconds
window_size_sec = 0.8
doc_plots.plot_history_window(neuron_count, epoch_one_spk, window_size_sec);
For each time point, we shift our window one bin at the time and vertically stack the spike count history in a matrix. Each row of the matrix will be used as the predictors for the rate in the next bin (red narrow rectangle in the figure).
doc_plots.run_animation(neuron_count, epoch_one_spk.start[0])
If \(t\) is smaller than the window size, we won’t have a full window of spike history for estimating the rate. One may think of padding the window (with zeros for example) but this may generate weird border artifacts. To avoid that, we can simply restrict our analysis to times \(t\) larger than the window and NaN-pad earlier time-points;
You can construct this feature matrix with the HistoryConv basis.
Question: Can you:
- Convert the window size in number of bins (call it window_size)
- Define an HistoryConv basis covering this window size (call it history_basis).
- Create the feature matrix with history_basis.compute_features (call it input_feature).
# convert the prediction window to bins (by multiplying with the sampling rate)
window_size = int(window_size_sec * neuron_count.rate)
# define the history bases
history_basis = nmo.basis.HistoryConv(window_size)
# create the feature matrix
input_feature = history_basis.compute_features(neuron_count)
NeMoS NaN pads if there aren’t enough samples to predict the counts.
# print the NaN indices along the time axis
print("NaN indices:\n", np.where(np.isnan(input_feature[:, 0]))[0])
The binned counts originally have shape “number of samples”, we should check that the dimension are matching our expectation
Question: Can you check the shape of the counts and features?
print(f"Time bins in counts: {neuron_count.shape[0]}")
print(f"Convolution window size in bins: {window_size}")
print(f"Feature shape: {input_feature.shape}")
print(f"Feature shape: {input_feature.shape}")
We can visualize the output for a few time bins
suptitle = "Input feature: Count History"
neuron_id = 0
workshop_utils.plot_features(input_feature, count.rate, suptitle);
As you may see, the time axis is backward, this happens because under the hood, the basis is using the convolution operator which flips the time axis. This is equivalent, as we can interpret the result as how much a spike will affect the future rate. In the previous tutorial our feature was 1-dimensional (just the current), now instead the feature dimension is 80, because our bin size was 0.01 sec and the window size is 0.8 sec. We can learn these weights by maximum likelihood by fitting a GLM.
Fitting the Model#
When working a real dataset, it is good practice to train your models on a chunk of the data and use the other chunk to assess the model performance. This process is known as “cross-validation”. There is no unique strategy on how to cross-validate your model; What works best depends on the characteristic of your data (time series or independent samples, presence or absence of trials…), and that of your model. Here, for simplicity use the first half of the wake epochs for training and the second half for testing. This is a reasonable choice if the statistics of the neural activity does not change during the course of the recording. We will learn about better cross-validation strategies with other examples.
# construct the train and test epochs
duration = input_feature.time_support.tot_length("s")
start = input_feature.time_support["start"]
end = input_feature.time_support["end"]
# define the interval sets
first_half = nap.IntervalSet(start, start + duration / 2)
second_half = nap.IntervalSet(start + duration / 2, end)
Question: Can you fit the glm to the first half of the recording and visualize the maximum likelihood weights?
# define the GLM object
model = nmo.glm.GLM(solver_name="LBFGS")
# Fit over the training epochs
model.fit(
input_feature.restrict(first_half),
neuron_count.restrict(first_half)
)
The weights represent the effect of a spike at time lag \(i\) on the rate at time \(t\). The next cell display the learned weights.
The model should be called model from the previous cell.
plt.figure()
plt.title("Spike History Weights")
plt.plot(np.arange(window_size) / count.rate, np.squeeze(model.coef_), lw=2, label="GLM raw history 1st Half")
plt.axhline(0, color="k", lw=0.5)
plt.xlabel("Time From Spike (sec)")
plt.ylabel("Kernel")
plt.legend()
The response in the previous figure seems noise added to a decay, therefore the response can be described with fewer degrees of freedom. In other words, it looks like we are using way too many weights to describe a simple response. If we are correct, what would happen if we re-fit the weights on the other half of the data?
Inspecting the results#
Question: Can you fit the model on the second half of the data and compare the results?
# fit on the other half of the data
model_second_half = nmo.glm.GLM(solver_name="LBFGS")
model_second_half.fit(
input_feature.restrict(second_half),
neuron_count.restrict(second_half)
)
Compare results.
plt.figure()
plt.title("Spike History Weights")
plt.plot(np.arange(window_size) / count.rate, np.squeeze(model.coef_),
label="GLM raw history 1st Half", lw=2)
plt.plot(np.arange(window_size) / count.rate, np.squeeze(model_second_half.coef_),
color="orange", label="GLM raw history 2nd Half", lw=2)
plt.axhline(0, color="k", lw=0.5)
plt.xlabel("Time From Spike (sec)")
plt.ylabel("Kernel")
plt.legend()
What can we conclude?
The fast fluctuations are inconsistent across fits, indicating that they are probably capturing noise, a phenomenon known as over-fitting; On the other hand, the decaying trend is fairly consistent, even if our estimate is noisy. You can imagine how things could get worst if we needed a finer temporal resolution, such 1ms time bins (which would require 800 coefficients instead of 80). What can we do to mitigate over-fitting now?
Reducing feature dimensionality#
Let’s see how to use NeMoS’ basis module to reduce dimensionality and avoid over-fitting!
For history-type inputs, we’ll use again the raised cosine log-stretched basis,
Pillow et al., 2005.
doc_plots.plot_basis();
We can initialize the RaisedCosineLogConv by providing the number of basis functions
and the window size for the convolution. With more basis functions, we’ll be able to represent
the effect of the corresponding input with the higher precision, at the cost of adding additional parameters.
Question: Can you define the basis RaisedCosineLogConvand name it basis?
Basis parameters:
8 basis functions.
Window size of 0.8sec.
# a basis object can be instantiated in "conv" mode for convolving the input.
basis = nmo.basis.RaisedCosineLogConv(
n_basis_funcs=8, window_size=window_size
)
Our spike history predictor was huge: every possible 80 time point chunk of the data, for \(144 \cdot 10^4\) total numbers. By using this basis set we can instead reduce the predictor to 8 numbers for every 80 time point window for \(144 \cdot 10^3\) total numbers, an order of magnitude less. With 1ms bins we would have achieved 2 order of magnitude reduction in input size. This is a huge benefit in terms of memory allocation and, computing time. As an additional benefit, we will reduce over-fitting.
Let’s see our basis in action. We can “compress” spike history feature by convolving the basis
with the counts (without creating the large spike history feature matrix).
This can be performed in NeMoS by calling the compute_features method of basis.
Question: Can you:
Convolve the counts with the basis functions. (Call the output
conv_spk)Print the shape of
conv_spkand compare it toinput_feature.
# equivalent to
# `nmo.convolve.create_convolutional_predictor(basis_kernels, neuron_count)`
conv_spk = basis.compute_features(neuron_count)
print(f"Raw count history as feature: {input_feature.shape}")
print(f"Compressed count history as feature: {conv_spk.shape}")
Let’s focus on two small time windows and visualize the features, which result from convolving the counts with the basis elements.
# Visualize the convolution results
epoch_one_spk = nap.IntervalSet(8917.5, 8918.5)
epoch_multi_spk = nap.IntervalSet(8979.2, 8980.2)
doc_plots.plot_convolved_counts(neuron_count, conv_spk, epoch_one_spk, epoch_multi_spk);
Fit and compare the models#
Now that we have our “compressed” history feature matrix, we can fit the ML parameters for a GLM.
Question: Can you fit the model using the compressed features? Call it model_basis.
# use restrict on interval set training
model_basis = nmo.glm.GLM(solver_name="LBFGS")
model_basis.fit(conv_spk.restrict(first_half), neuron_count.restrict(first_half))
We can plot the resulting response, noting that the weights we just learned needs to be “expanded” back
to the original window_size dimension by multiplying them with the basis kernels.
We have now 8 coefficients,
print(model_basis.coef_)
In order to get the response we need to multiply the coefficients by their corresponding basis function, and sum them.
Question: Can you:
Reconstruct the history filter:
Extract the basis kernels with
_, basis_kernels = basis.evaluate_on_grid(window_size).Multiply the
basis_kernelwith the coefficient usingnp.matmul.
Check the shape of
self_connection.
_, basis_kernels = ... # get the basis function kernels
self_connection = ... # multiply with the weights
print(self_connection.shape)
# get the basis function kernels
_, basis_kernels = basis.evaluate_on_grid(window_size)
# multiply with the weights
self_connection = np.matmul(basis_kernels, model_basis.coef_)
print(self_connection.shape)
Let’s check if our new estimate does a better job in terms of over-fitting. We can do that by visual comparison, as we did previously. Let’s fit the second half of the dataset.
Question: Can you fit the other half of the data. Name it model_basis_second_half.
model_basis_second_half = nmo.glm.GLM(solver_name="LBFGS").fit(
conv_spk.restrict(second_half), neuron_count.restrict(second_half)
)
Question: Can you:
Get the response filters? Multiply the
basis_kernelswith the weights frommodel_basis_second_half.Call the output
self_connection_second_half.
self_connection_second_half = np.matmul(basis_kernels, model_basis_second_half.coef_)
And plot the results.
time = np.arange(window_size) / count.rate
plt.figure()
plt.title("Spike History Weights")
plt.plot(time, np.squeeze(model.coef_), "k", alpha=0.3, label="GLM raw history 1st half")
plt.plot(time, np.squeeze(model_second_half.coef_), alpha=0.3, color="orange", label="GLM raw history 2nd half")
plt.plot(time, self_connection, "--k", lw=2, label="GLM basis 1st half")
plt.plot(time, self_connection_second_half, color="orange", lw=2, ls="--", label="GLM basis 2nd half")
plt.axhline(0, color="k", lw=0.5)
plt.xlabel("Time from spike (sec)")
plt.ylabel("Weight")
plt.legend()
Question: Can you:
Predict the rates from
modelandmodel_basis? Call itrate_historyandrate_basis.Convert the rate from spike/bin to spike/sec by multiplying with
conv_spk.rate?
rate_basis = model_basis.predict(conv_spk) * conv_spk.rate
rate_history = model.predict(input_feature) * conv_spk.rate
And plot it.
ep = nap.IntervalSet(start=8819.4, end=8821)
# plot the rates
doc_plots.plot_rates_and_smoothed_counts(
neuron_count,
{"Self-connection raw history":rate_history, "Self-connection bsais": rate_basis}
);
All-to-all Connectivity#
The same approach can be applied to the whole population. Now the firing rate of a neuron
is predicted not only by its own count history, but also by the rest of the
simultaneously recorded population. We can convolve the basis with the counts of each neuron
to get an array of predictors of shape, (num_time_points, num_neurons * num_basis_funcs).
Preparing the features#
Question: Can you:
Re-define the basis?
Convolve all counts? Call the output in
convolved_count.Print the output shape?
Since this time we are convolving more than one neuron, we need to reset the expected input shape.
This can be done by passing the population counts to the set_input_shape method.
basis = ... # reset the input shape by passing the population count
convolved_count = ... # convolve all the neurons
# reset the input shape by passing the pop. count
print(count.shape)
print(152/8)
basis.set_input_shape(count)
# convolve all the neurons
convolved_count = basis.compute_features(count)
Check the dimension to make sure it make sense.
Shape should be (n_samples, n_basis_func * n_neurons)
print(f"Convolved count shape: {convolved_count.shape}")
Fitting the Model#
This is an all-to-all neurons model.
We are using the class PopulationGLM to fit the whole population at once.
Once we condition on past activity, log-likelihood of the population is the sum of the log-likelihood
Question: Can you:
Fit a
PopulationGLM? Call the objectmodelUse Ridge regularization with a
regularizer_strength=0.1?Print the shape of the estimated coefficients.
model = nmo.glm.PopulationGLM(
regularizer="Ridge",
solver_name="LBFGS",
regularizer_strength=0.1
).fit(convolved_count, count)
print(f"Model coefficients shape: {model.coef_.shape}")
Comparing model predictions.#
Predict the rate (counts are already sorted by tuning prefs)
**Question: Can you:**11
Predict the firing rate of each neuron? Call it
predicted_firing_rate.Convert the rate from spike/bin to spike/sec?
predicted_firing_rate = ... # predict the rate from the model
predicted_firing_rate = model.predict(convolved_count) * conv_spk.rate
Plot fit predictions over a short window not used for training.
# use pynapple for time axis for all variables plotted for tick labels in imshow
workshop_utils.plot_head_direction_tuning_model(tuning_curves, spikes, angle,
predicted_firing_rate, threshold_hz=1,
start=8910, end=8960, cmap_label="hsv");
Let’s see if our firing rate predictions improved and in what sense.
fig = doc_plots.plot_rates_and_smoothed_counts(
neuron_count,
{"Self-connection: raw history": rate_history,
"Self-connection: bsais": rate_basis,
"All-to-all: basis": predicted_firing_rate[:, 0]}
)
Visualizing the connectivity#
Finally, we can extract and visualize the pairwise interactions between neurons.
**Question: Can you extract the weights and store it in a (n_neurons, n_neurons, n_basis_funcs) array?
# original shape of the weights
print(f"GLM coeff: {model.coef_.shape}")
You can use the split_by_feature method of basis for this.
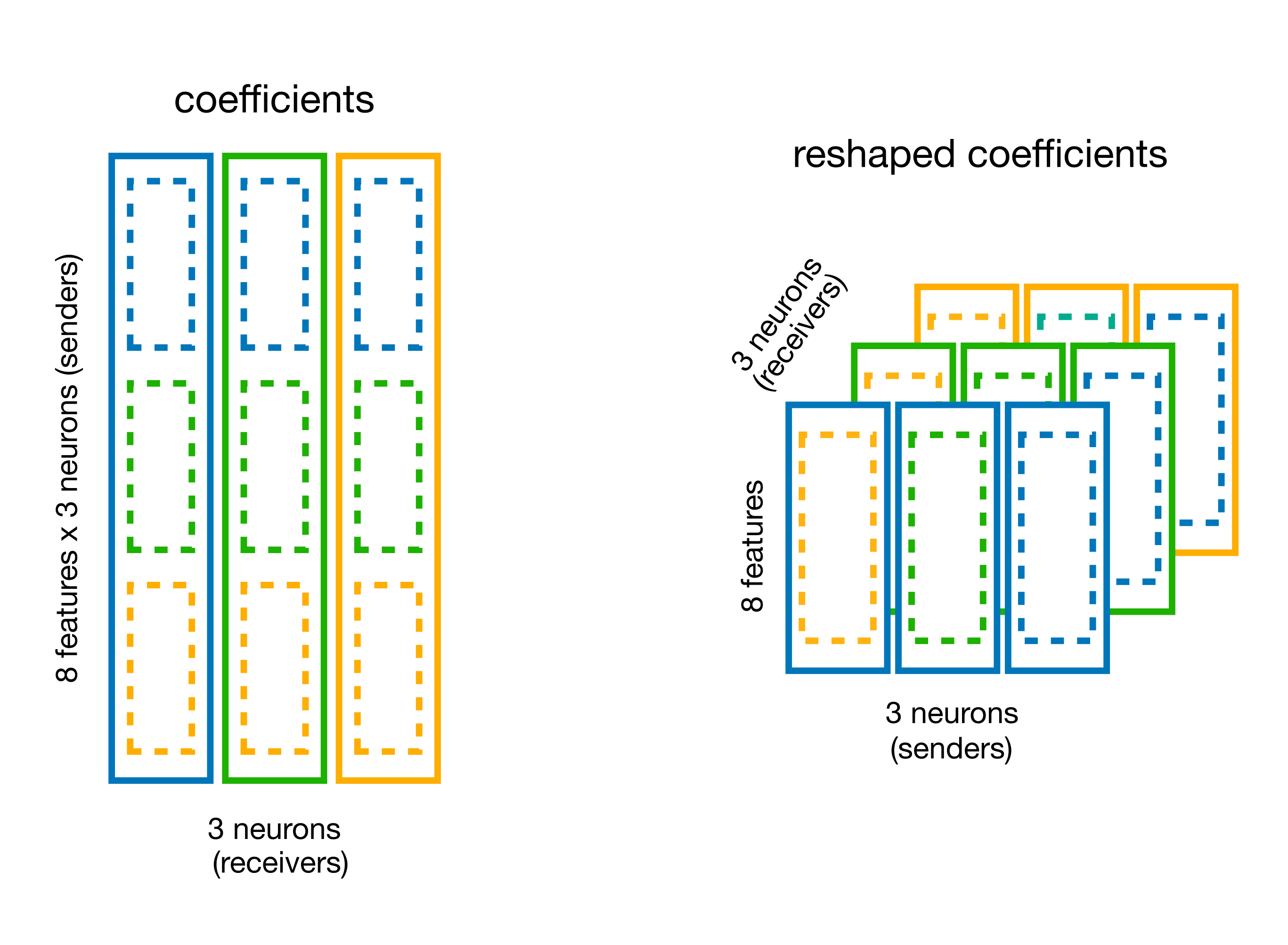
# split the coefficient vector along the feature axis (axis=0)
weights_dict = basis.split_by_feature(...)
# visualize the content
weights = weights_dict["RaisedCosineLogConv"]
# split the coefficient vector along the feature axis (axis=0)
weights_dict = basis.split_by_feature(model.coef_, axis=0)
# the output is a dict with key the basis label,
# and value the reshaped coefficients
weights = weights_dict["RaisedCosineLogConv"]
print(f"Re-shaped coeff: {weights.shape}")
The shape is (sender_neuron, num_basis, receiver_neuron).
Let’s reconstruct the coupling filters by multiplying the weights with the basis functions.
responses = np.einsum("jki,tk->ijt", weights, basis_kernels)
print(responses.shape)
Finally, we can visualize the pairwise interactions by plotting all the coupling filters.
predicted_tuning_curves = nap.compute_tuning_curves(
data=predicted_firing_rate,
features=angle,
bins=61,
epochs = angle.time_support,
range=(0, 2 * np.pi),
feature_names = ["Angle"]
)
fig = workshop_utils.plot_coupling_filters(responses, predicted_tuning_curves)